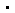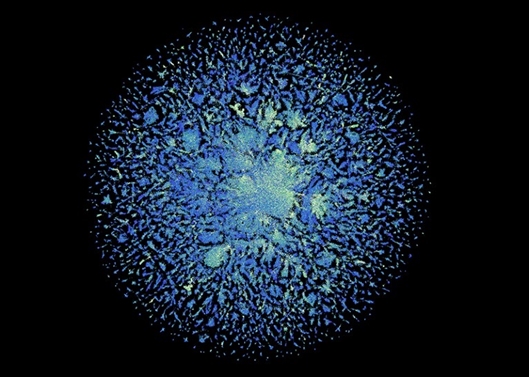
esm宏基因组图谱数据库包含6.17亿个蛋白质的结构预测。 图片来源:esm宏基因组图谱
谷歌旗下人工智能(ai)公司deep mind今年公布了2.2亿个蛋白质的预测结构,几乎涵盖了dna数据库中已知生物的所有蛋白质。现在,另一个科技巨头正在填补蛋白质宇宙中的暗物质。
meta公司(前身为facebook)的研究人员使用人工智能预测了约6亿个蛋白质的结构,这些蛋白质来自细菌、病毒和其他尚未被表征的微生物。相关研究11月1日发表于预印本网站biorxiv。
“这些是非常神秘的蛋白质,为深入了解生物学提供了可能性。”meta人工智能蛋白质团队研究负责人alexander rives说。
该团队使用“大型语言模型”生成了这些预测。“大型语言模型”是一种人工智能,可作为通过几个字母或单词预测文本的工具的基础。
通常语言模型是在大量文本的基础上进行训练的。为了将其应用于蛋白质,rives团队将已知蛋白质序列“喂”给它们,这些蛋白质可以由20个不同的氨基酸链表示,每个氨基酸链由一个字母表示。然后,该模型学会了在氨基酸比例模糊的情况下“自动补全”蛋白质。
rives说,这种训练使模型对蛋白质序列有了直观的理解,蛋白质序列包含了蛋白质形状的信息。
第二步,受deepmind开创性蛋白质结构人工智能算法alphafold的启发,模型将这种洞察力与已知蛋白质结构和序列之间关系的信息相结合,从蛋白质序列中生成预测结构。
今年夏天早些时候,rives团队报告称,其模型算法名为esmfold,虽准确性不如alphafold,但在预测结构方面要快60倍左右。“这意味着我们可以将结构预测扩展到更大的数据库中。”rives说。
作为一个测试案例,研究团队决定将模型应用于大规模测序的“宏基因组”dna数据库,这些dna来自于环境,包括土壤、海水、人类肠道、皮肤和其他微生物栖息地。绝大多数编码潜在蛋白质的dna条目来自从未被培养过的生物,也不为科学家所知。
meta团队总共预测了超过6.17亿个蛋白质的结构,这项工作只花了两周时间。rives表示,预测是免费的,任何人都可以使用,就像模型的底层代码一样。
在这6.17亿个预测中,该模型认为超过1/3的预测是高质量的,因此研究人员可以确信蛋白质的整体形状是正确的,在某些情况下,模型可以识别更精细的原子级细节。值得一提的是,其中数以百万计的结构都是全新的,与实验确定的蛋白质结构数据库,或从已知生物体预测的alphafold数据库中的结构都不同。
alphafold数据库的很大一部分是由彼此几乎相同的结构组成的,而宏基因组数据库则应该涵盖以前从未见过的蛋白质宇宙的很大一部分。
美国哈佛大学进化生物学家sergey ovchinnikov对esmfold做出的数亿个预测表示怀疑。他认为,有些蛋白质可能缺乏确定的结构,而另一些可能是非编码dna,被误认为是蛋白质编码材料。
德国慕尼黑工业大学计算生物学家burkhard rost对meta公司模型的速度和准确性的结合印象深刻。但他质疑,从宏基因组数据库预测蛋白质是否真的比alphafold的精确度更高。基于语言模型的预测方法,更适合快速确定突变如何改变蛋白质结构,这是alphafold无法做到的。
据deepmind的一位代表说,该公司目前没有计划在其数据库中进行宏基因组结构预测,但不排除在未来这样做的可能性。
韩国首尔国立大学计算生物学家martin steinegger认为,这类工具的下一步显然是研究生物学中的暗物质。“我们很快就会在这些宏基因组结构的分析方面出现爆炸式增长。”(来源:中国科学报 辛雨)
相关论文信息: