2021年10月22日,山东师范大学的唐波教授与北京大学深圳研究生院的周建荣研究院课题组合作在angew. chem. int. ed.上发表了一篇题为 “enantioselective synthesis of chiral carboxylic acids from alkynes and formic acid by nickel-catalyzed cascade reactions: facile synthesis of profens” 的新研究。
课题组利用廉价金属镍催化炔烃连续发生氢羧基化和不对称转移氢化反应,一步法合成了手性羧酸,其中甲酸既是羧基化的碳源也是氢化反应的氢源。该串联反应成功地应用于布洛芬、萘普生、氟比洛芬三种重要的丙酸类非甾体抗炎药的不对称合成。
山东师范大学青年教师杨朋副教授和硕士研究生孙雅鑫为共同第一作者。
手性羧酸及其衍生物是药物分子中常见的重要结构骨架。除了手性拆分法,不对称催化合成手性羧酸的方法主要有两种:第一种方法是烯烃的不对称氢羧基化(或氢酯化)反应,该反应一般需要使用高压有毒的co气体。虽然甲酸也可作为co的替代物,但是目前尚未实现不对称氢羧基化;第二种方法是过渡金属或酶催化的α,β-不饱和羧酸及其衍生物的不对称氢化反应,该方法已经成功应用到工业生产中,但是原料α,β-不饱和羧酸一般需要多步合成,使生产成本较高。近年来报道的过渡金属催化炔烃的氢羧基化反应可以一步合成α,β-不饱和羧酸,其中co、co2和hco2h是常见的碳一来源。
如果把氢羧基化反应和不对称氢化反应合并在一锅法完成,由简单炔烃直接合成手性羧酸,将是目前布洛芬、萘普生、氟比洛芬等临床广泛应用的丙酸类镇痛抗炎药物最简短的制备方法之一。这个设计的难度在于:一个催化剂能够催化两种完全不同类型的反应,反应速率需同步并且生成手性产物。

图1:串联反应示意图
首先,作者以苯乙炔(1a)为模型底物,经过大量筛选,找到了适合金属镍的最佳的双膦配体(r, r)-benzp*。以镍-benzp*原位生成的配合物为催化剂,经过优化,在1,4-二氧六环中添加0.3当量乙酸酐ac2o,且甲酸和三乙胺的比例为6:2时,能够以80%产率和88% ee得到目标产物。在确定了最优反应条件后,作者拓展了反应底物的范围(图2)。结果表明,各种类型芳基乙炔,杂环芳基乙炔以及长链脂肪端炔均能以优异的产率和对映选择性转化为相应手性酸。
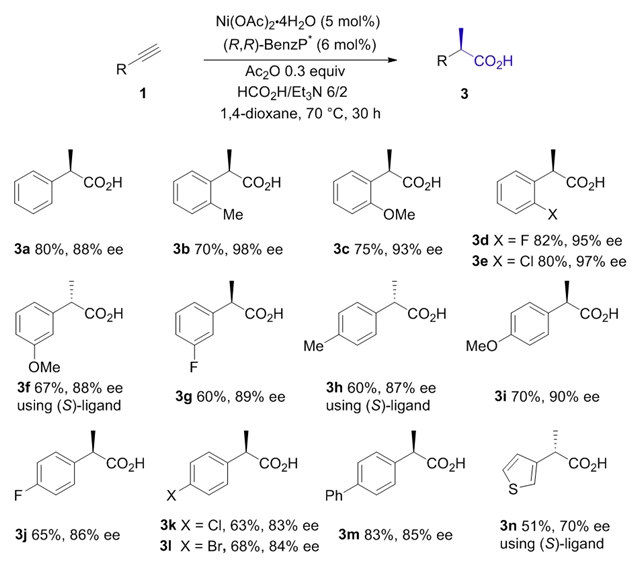
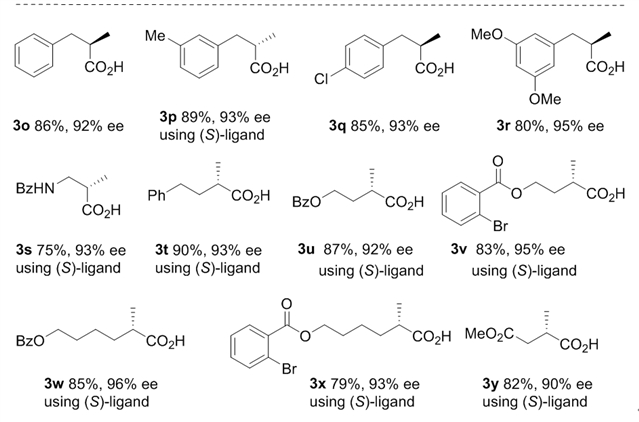
图2:底物拓展
随后,作者使用此方法成功合成到了布洛芬、萘普生、氟比洛芬三种药物分子,并且实现了克级规模的氟比洛芬合成(图3)。
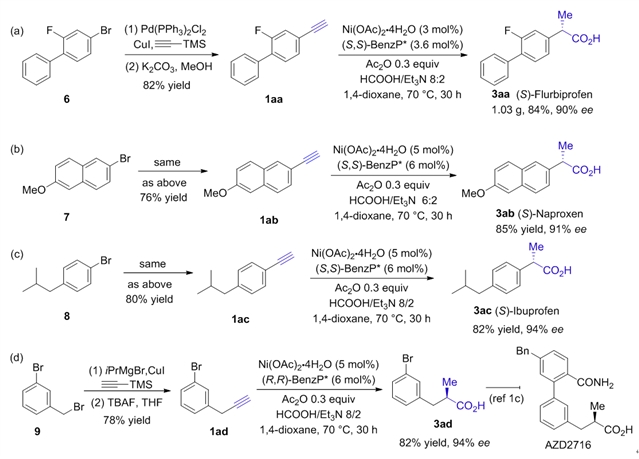
图3:药物合成应用
最后,作者通过氘代实验对该反应的机理进一步研究(图4)。发现产品中α-位的氢来自于甲酸的c-h,而β-位的氢来自于甲酸的o-h。据此,提出了一个可能的反应历程:在羧基化步骤,催化量乙酸酐与甲酸反应生成co,镍-炔络合物经质子化、co插入和还原消除生成丙烯酸,在转移氢化步骤,甲酸与镍络合,然后释放co2生成ni-h物种,ni-h插入到c=c键,随后被h net3质子化,得到最终产物。手性发生在ni-h插入步骤中,三乙胺发挥了质子梭的作用,一方面结合了甲酸中的质子,使甲酸根离子更容易与镍催化剂配位,另一方面影响了中间体(i)的质子化速率和立体化学。
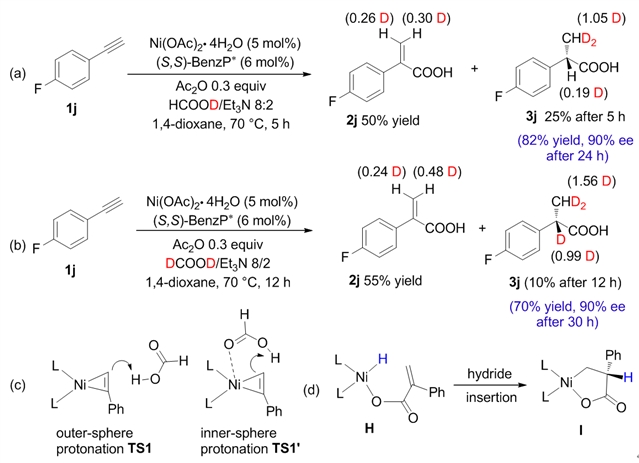
图4:氘代实验研究反应机理
随着社会对化工生产安全性和绿色性的要求不断提高,设计并实现更高效的一锅法绿色串联反应,可以缩短医药、农药等化学品的合成步骤,节约生产成本,具有重要的经济价值和学术意义。本项目得到了国家自然科学基金、山东省自然科学基金、山东师范大学、北京大学深圳研究生院和深圳湾实验室的大力支持。(来源:科学网)
相关论文信息: